



On February 23, 2026, a research team from the State Key Laboratory of Genome and Multi-omics Technologies at BGI-Research reports in Nature Communications an end-to-end nanopore sensing workflow designed to profile peptides and support protein identification at high throughput. By combining a streamlined approach for building “oligo–peptide–oligo” (OPO) libraries from native peptides or protein digests with an AI-driven analytical pipeline, the study addresses several persistent practical barriers in nanopore-based proteomics: preparing diverse samples in a broadly compatible format, acquiring sufficient single-molecule events in parallel, and extracting reliable information from electrical signals with noise.
 The study “Nanopore-based massively parallel sensing for peptide profiling and protein identification” was published in Nature Communications.
The study “Nanopore-based massively parallel sensing for peptide profiling and protein identification” was published in Nature Communications.
In the reported workflow, proteins are enzymatically fragmented into peptides and chemically coupled to DNA handles at both termini, forming an OPO “sandwich” structure with built-in signal markers that allow algorithms to automatically locate and extract the peptide-specific blockade from each current trace. Using a prototype 256-channel microwell array with CsgG nanopores, the team collected over 100,000 translocation events per library within a two-hour run, making population-level analysis of translocation events feasible rather than relying on sparse single-channel reads.
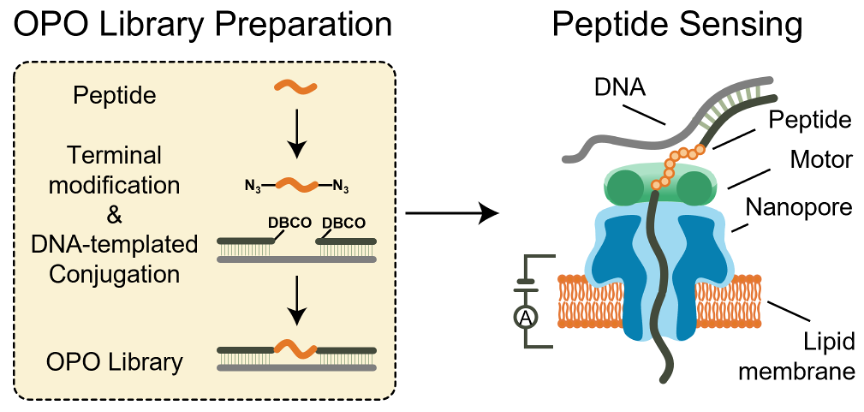
Bridging native proteins and sequencing hardware: attaching DNA handles to peptide fragments creates an Oligo-Peptide-Oligo (OPO) library that can be controllably driven through a nanopore by a motor protein for high-throughput electrical sensing.
A key insight is that individual peptide traces are highly variable unrepresentative, but aggregating thousands of high-quality events reveals reproducible, peptide-specific statistical pattern. The team captured these patterns in a “density matrix” fingerprint, which was then integrated with a convolutional neural network (CNN) classifier, using the density matrix as a secondary filter to reject low-confidence predictions. This combined strategy improved classification accuracy from 97.5% to 99.2% on a three-peptide benchmark, underscoring the critical role of the density matrix in capturing discriminative temporal dynamics.
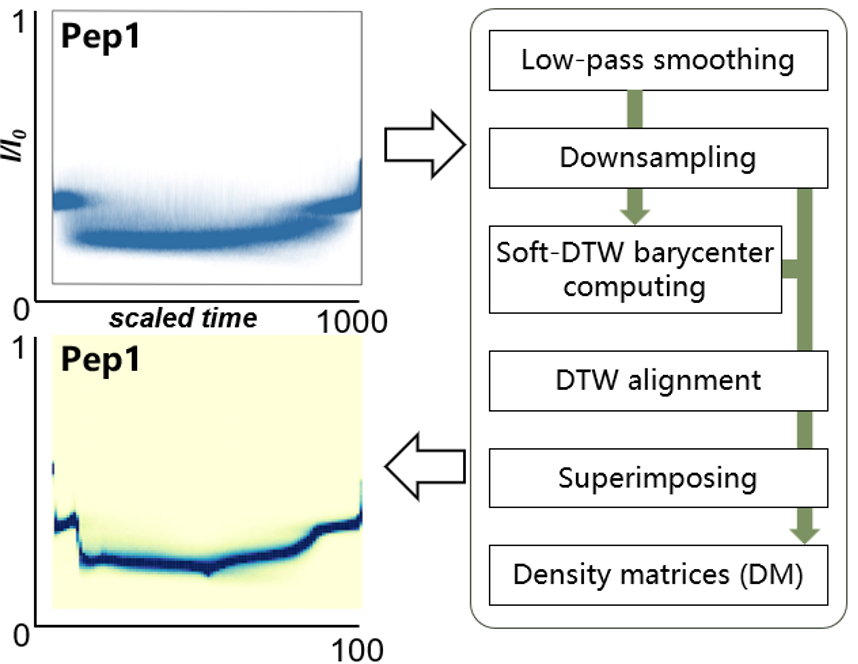
Extracting representatives from signals with microscopic fluctuations: an automated algorithmic workflow aligns and overlays thousands of highly variable peptide translocation signals to generate robust, visually distinct density matrices for highly accurate AI classification.
Building on this analytical foundation, the researchers tested how sensitively the platform can resolve subtle peptide differences. Using libraries that differ by single amino-acid variants, positional variants and post-translational modifications, the study shows that changes in charge and side-chain volume produce statistically distinguishable signal fingerprints, although certain especially challenging distinctions, such as leucine versus isoleucine isomers, can be clearly distinguished.
The team then applied this capability to validate and compare the binding affinities of various peptides for a specific antibody. By sequencing all peptides enriched by an antibody and distinguishing the peptide origins of all signals, the platform enabled rapid epitope mapping and antibody-pair compatibility assessment. In a separate demonstration using the FLAG-tag system, relative read abundances after enrichment tracked reported binding trends for several peptide variants, supporting semi-quantitative affinity estimation under the study’s experimental conditions.
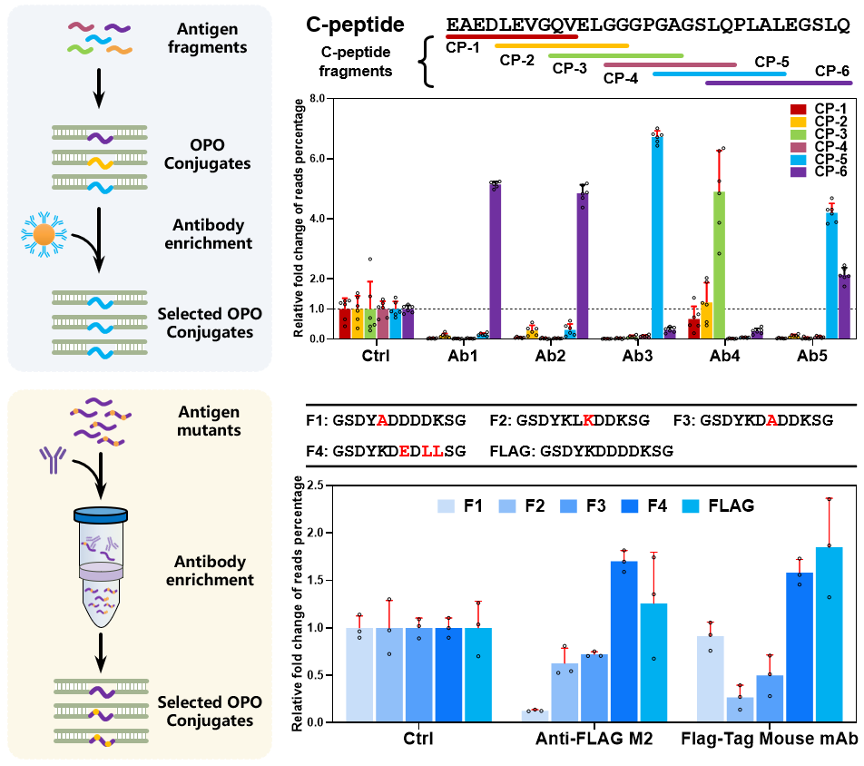
A digital readout for antibody validation: measuring the precise abundance of captured OPO-conjugated peptides after enrichment enables rapid, multiplexed mapping of binding epitopes and semi-quantitative assessment of antibody affinity.
Beyond peptide-level profiling, the authors also report a single-blind protein identification experiment in which LysC digests from three model proteins were analyzed against trained reference fingerprints, and the resulting peptide-distribution patterns enabled unambiguous assignment of the anonymized samples to their corresponding protein identities in that limited candidate set.
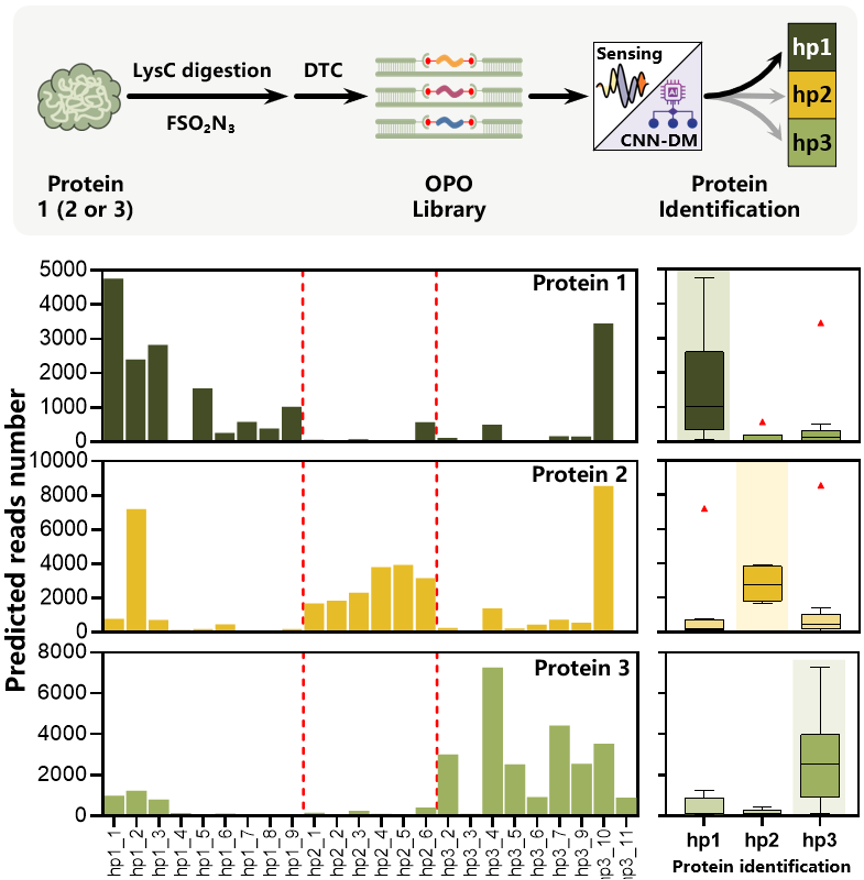
Unambiguous native protein identification: mapping the complex peptide read distributions from unknown enzymatic digests against pre-trained AI models successfully and blindly identifies whole proteins based on their multi-peptide signatures.
The authors emphasize that the work represents an integrated, scalable pipeline that jointly tackles limited throughput, native sample preparation and signal decoding. Looking ahead, Dr. Xun Xu, corresponding author, Chief Researcher of BGI Group and Director of State Key Laboratory of Genome and Multi-omics Technologies, notes that the team will focus on expanding protein fingerprint analyses toward higher-throughput affinity screening for drug discovery, while exploring targeted enrichment strategies to broaden the platform’s multi-omics potential.
This research is available at: https://doi.org/10.1038/s41467-026-69628-1