



Recently, a multi-institutional team led by BGI-Research, together with Dalian University of Technology and other collaborators, published a study in Nature Communications titled: "The Extreme Environment Microbiome Catalog (EEMC): a global resource for microbial diversity and antimicrobial discovery." The study reconstructs 78,213 microbial genomes from metagenomes and isolates across seven extreme habitats: deep-sea, cryosphere, subsurface, hypersaline, hyperacid, terrestrial geothermal, and hyperarid environments. Leveraging this global resource, the team further explores candidate antimicrobial peptides using protein language models, opening new avenues for antimicrobial discovery.
 The study “The Extreme Environment Microbiome Catalog (EEMC): a global resource for microbial diversity and antimicrobial discovery” was published in Nature Communications.
The study “The Extreme Environment Microbiome Catalog (EEMC): a global resource for microbial diversity and antimicrobial discovery” was published in Nature Communications.
Antimicrobial resistance is projected to contribute to roughly 39.1 million deaths between 2025 and 2050, yet no new class of antibiotics has been discovered since the 1980s. Microbes that thrive under extreme conditions, such as high salinity, temperature, pressure, and acidity, have long been considered a promising source of novel chemistry. However, past efforts have largely examined these systems in isolation. The EEMC was built to close this gap.
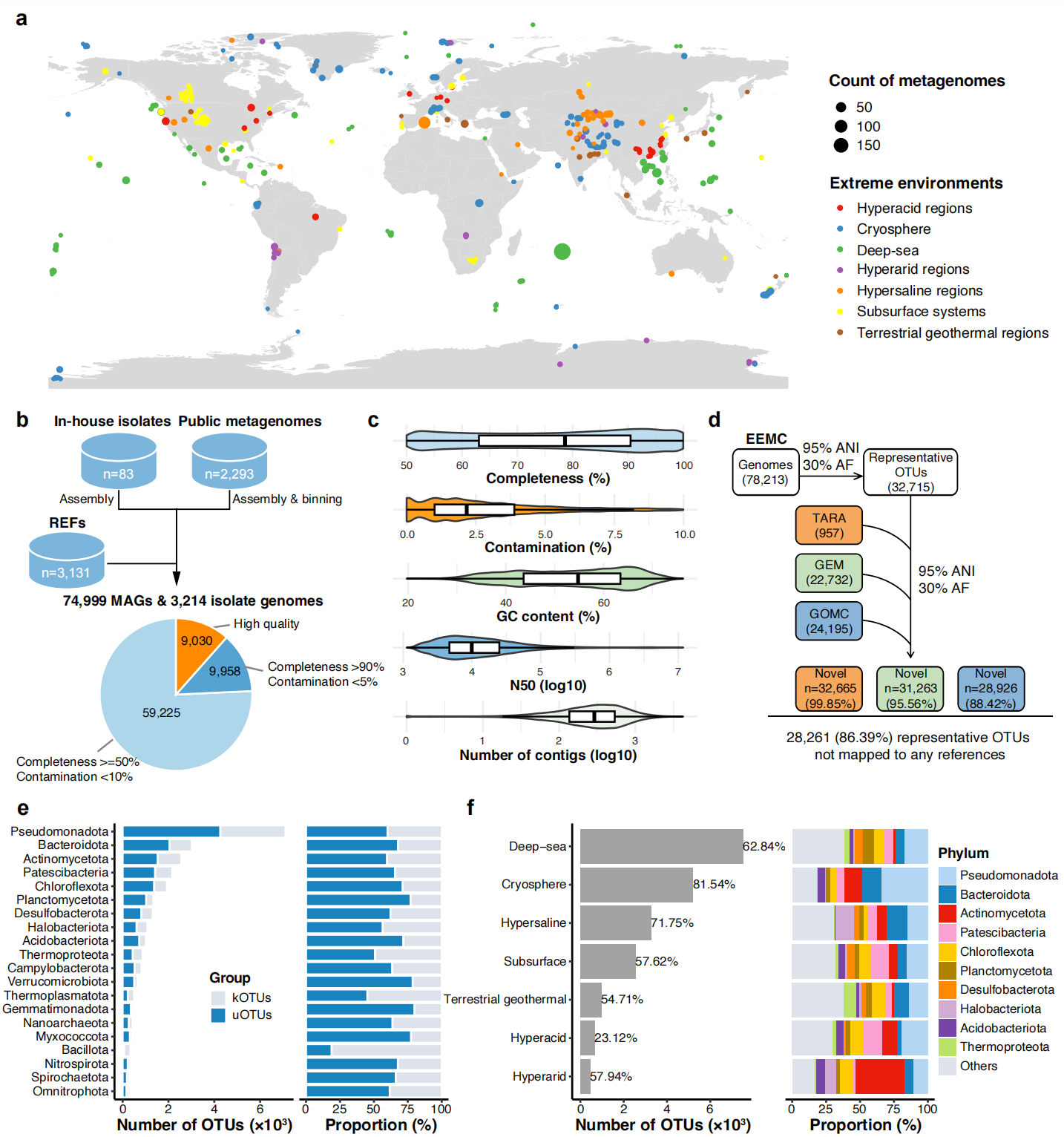 Metagenomes and isolates from seven extreme habitat categories were integrated into a global catalog, enabling large-scale cross-ecosystem comparisons of microbial diversity.
Metagenomes and isolates from seven extreme habitat categories were integrated into a global catalog, enabling large-scale cross-ecosystem comparisons of microbial diversity.
The team integrated 2,293 publicly available metagenomes with 3,214 cultured isolate genomes, including 83 strains newly isolated from Haima cold seep sediments in the northern South China Sea and sequenced on the DNBSEQ-T7 platform. After quality filtering, the catalog contains 78,213 bacterial and archaeal genomes that cluster into 32,715 species-level clusters. Notably, 20,610 species lack have no match in the Genome Taxonomy Database, and over 86.39% are missing from three widely used microbial catalogs, highlighting the vast unexplored microbial diversity captured in the EEMC. The accompanying gene set comprises nearly 4 billion non-redundant sequences, about one-fifth of which remain unannotated in standard functional databases, pointing to a substantial reservoir of unknown biological functions.
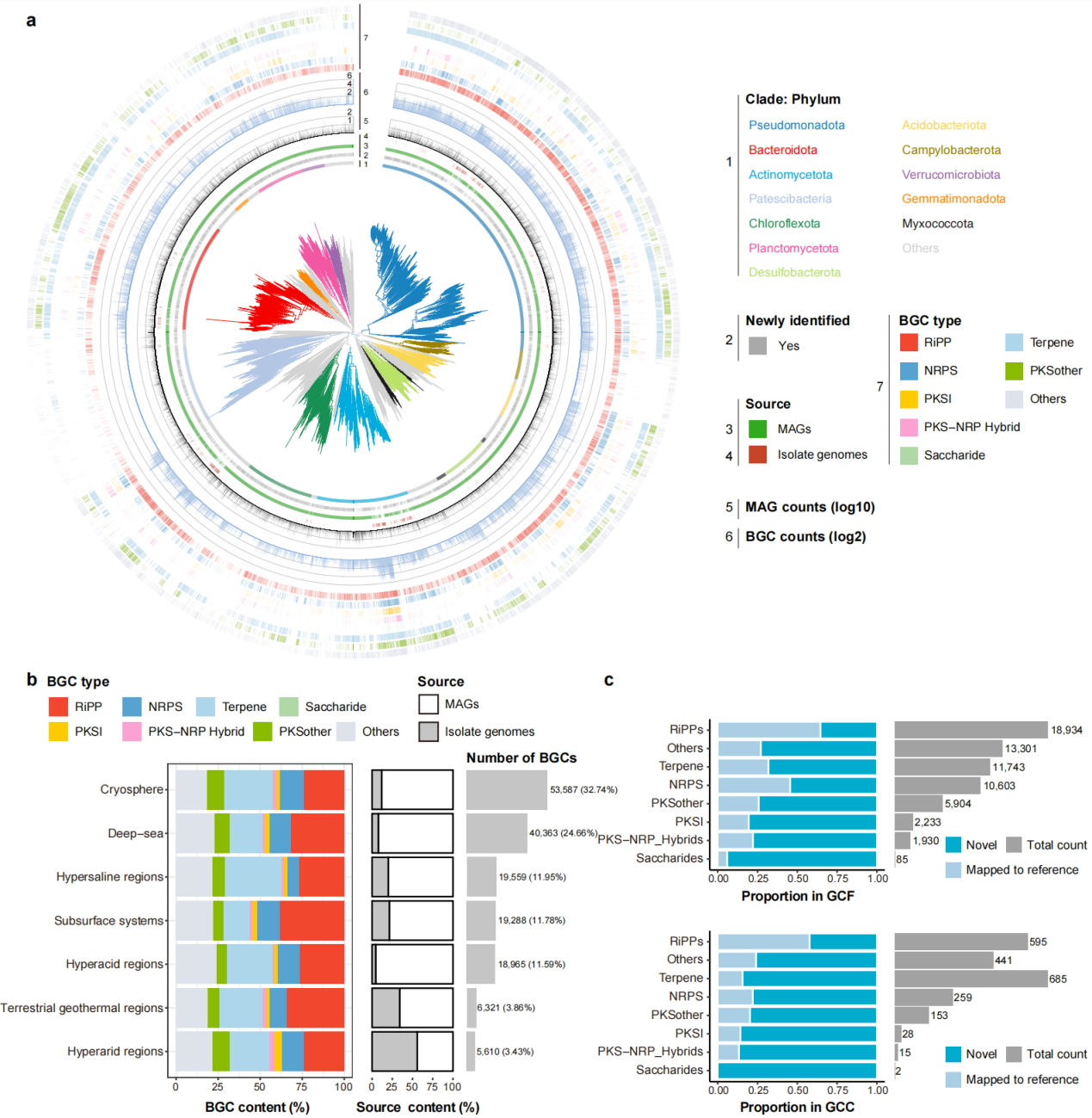 Extreme-environment microbes exhibit extensive biosynthetic diversity, with over half of gene cluster families classified as novel.
Extreme-environment microbes exhibit extensive biosynthetic diversity, with over half of gene cluster families classified as novel.
Using antiSMASH, the team identified 163,693 biosynthetic gene clusters (BGCs)—genomic regions that encode specialized metabolites, including many known classes of antibiotics.. These clusters were grouped into 64,733 families, with over half showing no similarity to known references, pointing to a vast reservoir of uncharted chemistry. Among the BGCs, ribosomally synthesized and post-translationally modified peptides (RiPPs) are of particular interest, as they are well-established as a rich source of antimicrobial peptides. They account for 28.60% of all identified clusters. Archaea further expand this landscape: 99.72% of archaeal BGC families are lineage-specific, revealing a largely unexplored biosynthetic space with potential implications for future antibiotic discovery.
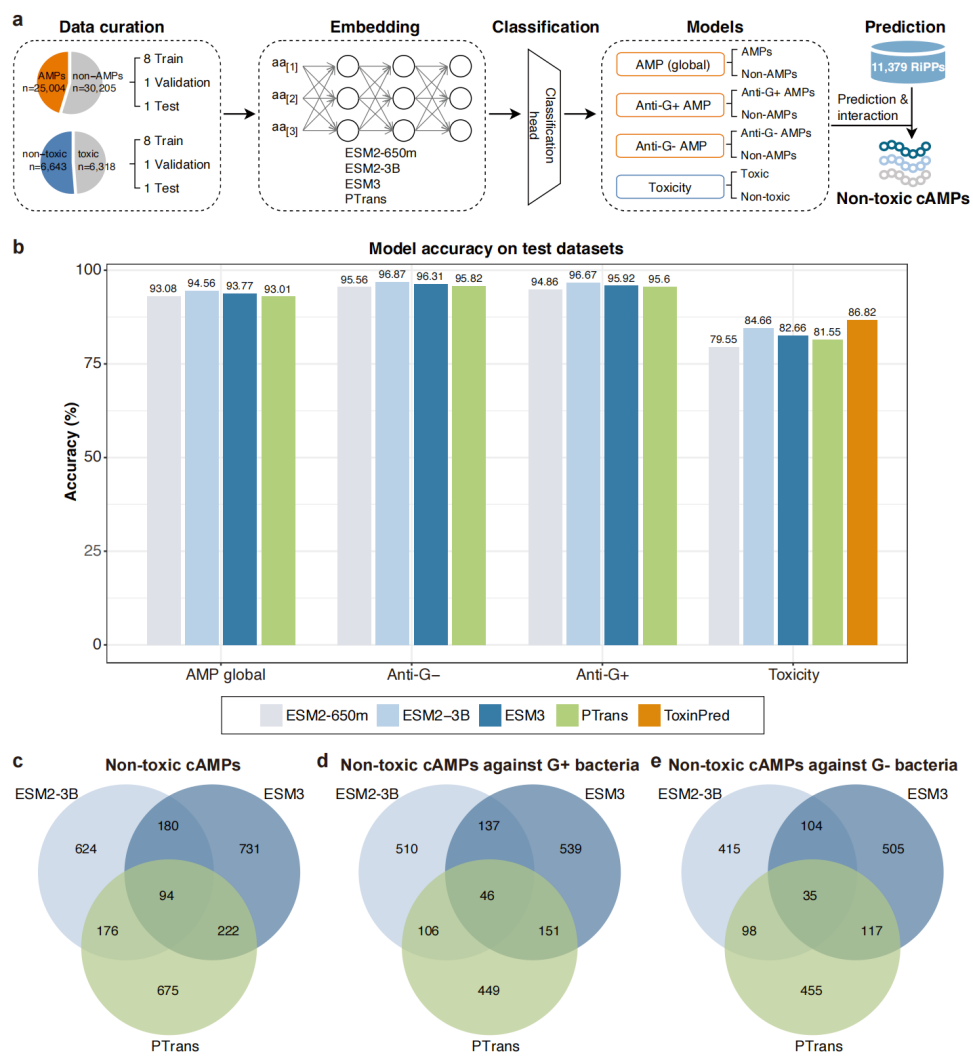 The MAI framework integrates protein large language models and downstream classifiers to prioritize peptides with antimicrobial activity and low mammalian toxicity.
The MAI framework integrates protein large language models and downstream classifiers to prioritize peptides with antimicrobial activity and low mammalian toxicity.
To translate this genetic potential into testable molecules, the team developed a framework called Metagenomics-AI (MAI). MAI leverages protein large language models to jointly evaluate peptide sequences for antimicrobial activity and mammalian toxicity, addressing a key limitation in peptide discovery: many candidates fail due to toxicity rather than lack of potency. Applied to 11,379 peptide sequences extracted from RiPP gene clusters, MAI prioritized 3,032 candidates predicted to be both active and non-toxic. The largest model tested, ESM2 with 3 billion parameters, outperformed established antimicrobial peptide predictors by more than 10 percentage points. Notably, most shortlisted peptides show low sequence similarity to the training data, suggesting strong potential for novel chemical space exploration.
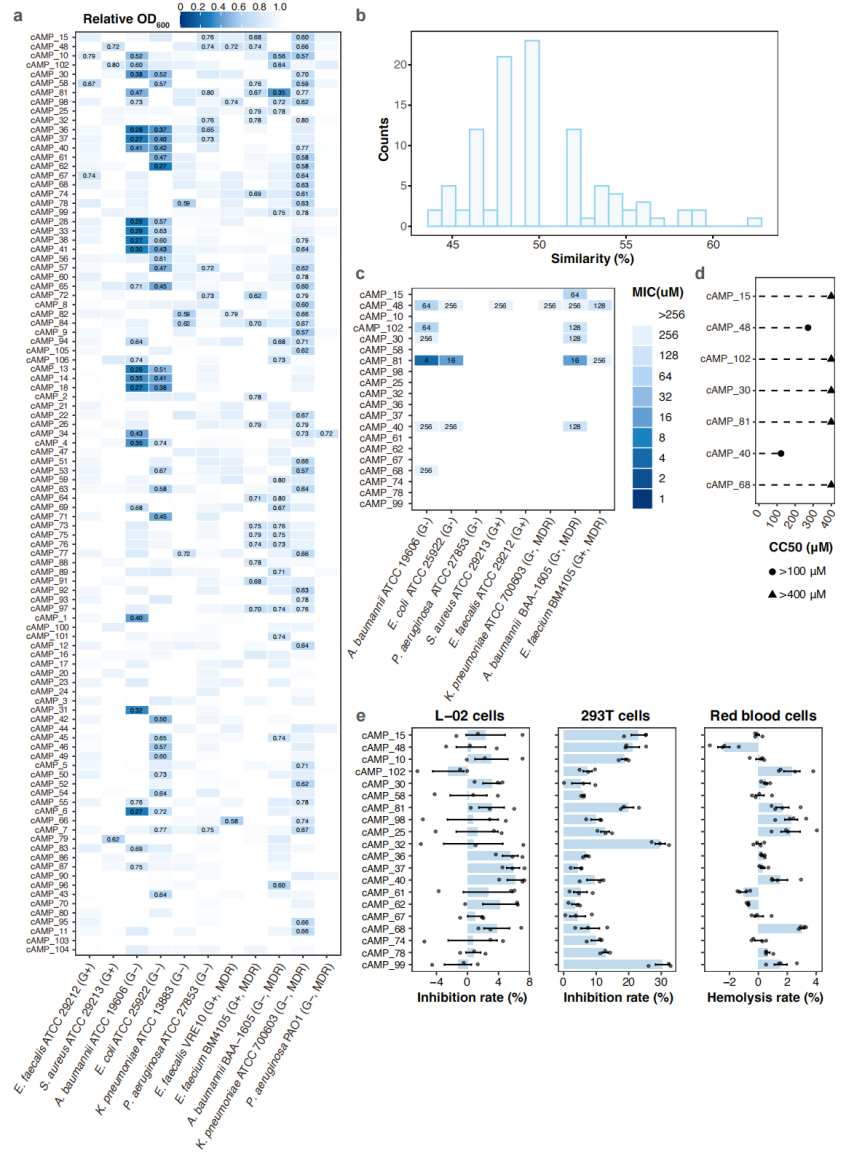
Experimental testing showed that several AI-prioritized peptides inhibited clinically relevant bacteria while exhibiting low toxicity in the assays performed.
To validate the computational predictions, the team synthesized 100 shortlisted peptides and tested them against 11 bacterial strains, including several multidrug-resistant pathogens. Among them, 84 peptides inhibited at least one strain, while toxicity assays showed generally low hemolysis and low cytotoxicity in human cell lines. One candidate, cAMP_81, showed particularly strong activity against Gram-negative pathogens, with minimum inhibitory concentrations as low as 4 μM against Acinetobacter baumannii and 16 μM against both Escherichia coli and a multidrug-resistant A. baumannii strain, comparable to or better than published positive controls. Mechanistic studies, including electron microscopy and membrane integrity assays, suggested that cAMP_81 primarily acts by disrupting the bacterial cytoplasmic membrane. Importantly, bacteria exposed to sublethal daily doses over 30 days developed less resistance than those treated with polymyxin B, a last-resort antibiotic. These results were obtained from in vitro assays of unmodified precursor peptides and do not constitute clinical evidence.
Beyond antibiotic discovery, the catalog provides a reusable reference for studying extremophile adaptation, identifying industrial enzymes, and expanding antimicrobial screening efforts. Looking forward, the team will integrate long-read sequencing, AI-based structure prediction, and synthetic biology to further explore extremophile-derived enzymes and bioactive metabolites. All data resources, including 74,999 metagenome-assembled genomes, 83 in-house isolate genomes, non-redundant gene sets, and 163,693 biosynthetic gene clusters, are publicly available in CNGBdb under accession CNP0007106. The MAI model weights are hosted on Zenodo, and inference scripts are available on GitHub.
The full paper is open access at https://doi.org/10.1038/s41467-026-71145-0.