



A research team led by BGI-Research, in collaboration with Peking Union Medical College Hospital and other institutions, publishes in Nature Genetics on June 11 the most comprehensive genomic atlas of the human vaginal microbiome to date. The study presents the Global Vaginal Metagenome-Assembled Genomes (GVMG) catalog: 65,055 microbial genomes from 13,632 metagenomic samples, capturing 890 prokaryotic species, 6,577 viral taxonomic units and 11 fungal species, many not previously represented in public databases. Furthermore, the researchers conducted the largest -to-date metagenome-genome-wide association study (M-GWAS) in this field, identifying seven host genetic loci associated with specific vaginal microbes.
 The study, publishing in Nature Genetics on June 11, 2026, presents the most comprehensive genomic atlas of the human vaginal microbiome to date.
The study, publishing in Nature Genetics on June 11, 2026, presents the most comprehensive genomic atlas of the human vaginal microbiome to date.
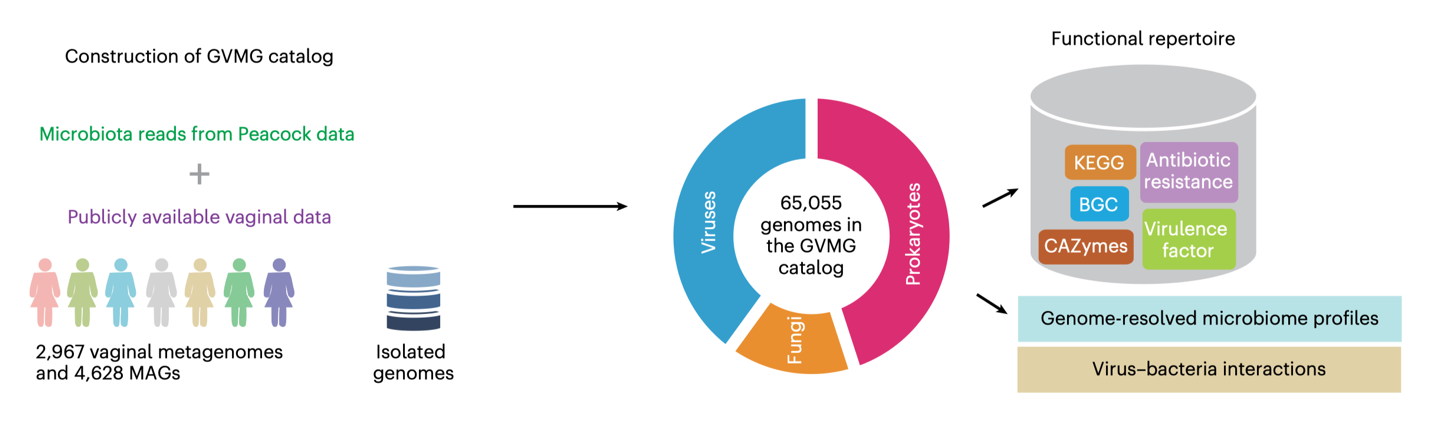 Construction of the GVMG catalog, integrating over 13,000 metagenomes from the Peacock cohort and public datasets into 65,055 microbial genomes spanning bacteria, viruses and fungi, nearly doubling previous vaginal microbiome genomic resources.
Construction of the GVMG catalog, integrating over 13,000 metagenomes from the Peacock cohort and public datasets into 65,055 microbial genomes spanning bacteria, viruses and fungi, nearly doubling previous vaginal microbiome genomic resources.
Prior vaginal microbiome studies offered limited species-level resolution and largely excluded viruses and fungi, while existing reference databases, built predominantly from Western populations, left a significant gap for Asian women and broader clinical contexts.
To address these limitations, the team assembled the Peacock cohort of over 10,000 cervicovaginal samples from Chinese women spanning a broad age and clinical spectrum, including healthy individuals and those with conditions such as bacterial vaginosis (BV) and HPV infection. Combined with approximately 3,000 publicly available metagenomes from the United States, France and other countries, the dataset was analyzed using shotgun metagenomic sequencing to achieve species-level and strain-level resolution across bacteria, viruses and fungi.
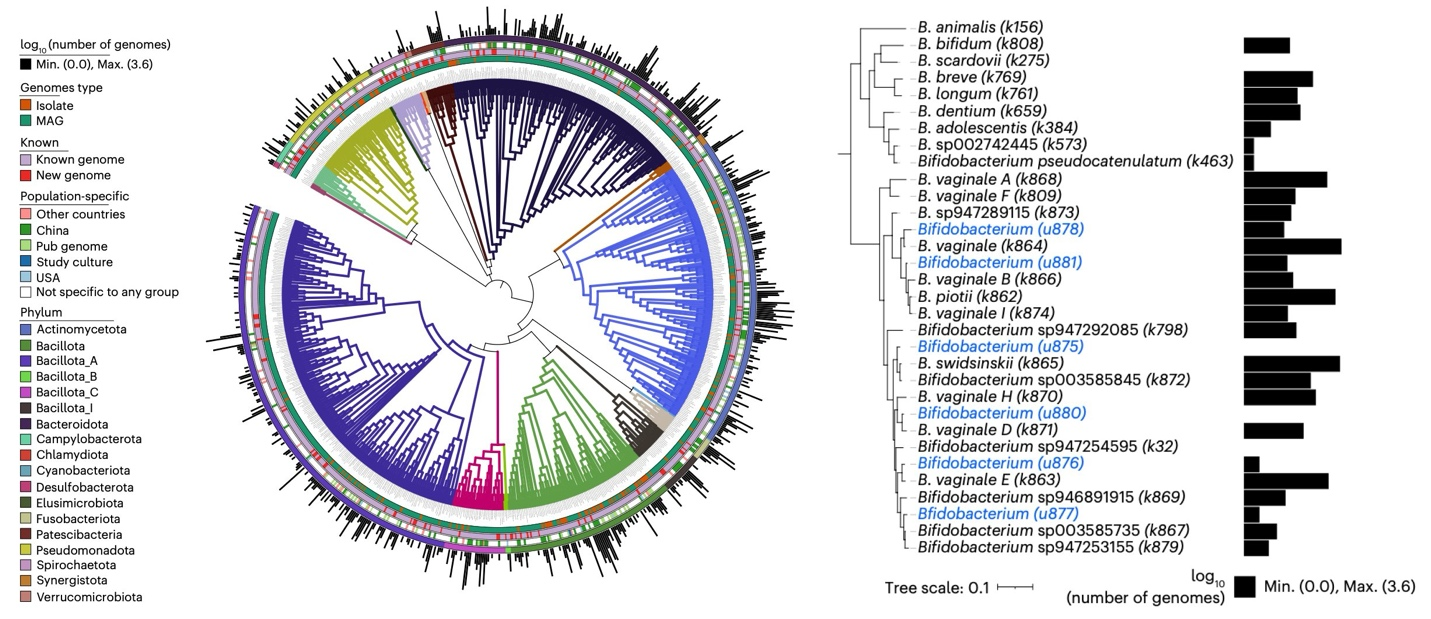 Phylogenetic diversity of 889 vaginal bacterial species in the GVMG catalog (left), with a detailed view of the Bifidobacterium genus (right) revealing 23 distinct genomospecies within B. vaginale, formerly classified as Gardnerella vaginalis.
Phylogenetic diversity of 889 vaginal bacterial species in the GVMG catalog (left), with a detailed view of the Bifidobacterium genus (right) revealing 23 distinct genomospecies within B. vaginale, formerly classified as Gardnerella vaginalis.
Among the catalog's findings, 13% of prokaryotic species and 79% of viral taxonomic units had not been recorded in existing public databases, underscoring the extent of previously uncharacterized microbial diversity in the vaginal ecosystem. One notable result was the identification of 23 distinct genomospecies within what was long considered a single species, Gardnerella vaginalis (now reclassified as Bifidobacterium vaginale under the Genome Taxonomy Database). These genomospecies differ in their carriage of sialidases and cytolysins, enzymes that can degrade protective mucus and damage epithelial cells, suggesting that BV involves a more varied set of organisms than previously appreciated.
Across the broader catalog, 96% of Lactobacillus iners genomes carried cytolysin genes, suggesting that this common species may compromise the mucosal barrier under certain conditions, whereas Lactobacillus crispatus showed minimal sialidase or cytolysin activity, reinforcing its recognized role as the primary protective species in the vaginal ecosystem.
The study also revealed pronounced population-specific genomic variation at the strain level. BVAB1, a bacterium increasingly linked to vaginal dysbiosis and preterm birth, was detected in 12.4% of samples from the American cohort but only 0.46% of Chinese samples, and American-origin strains harbored a higher prevalence of virulence-associated biosynthetic gene clusters at the genomic level.
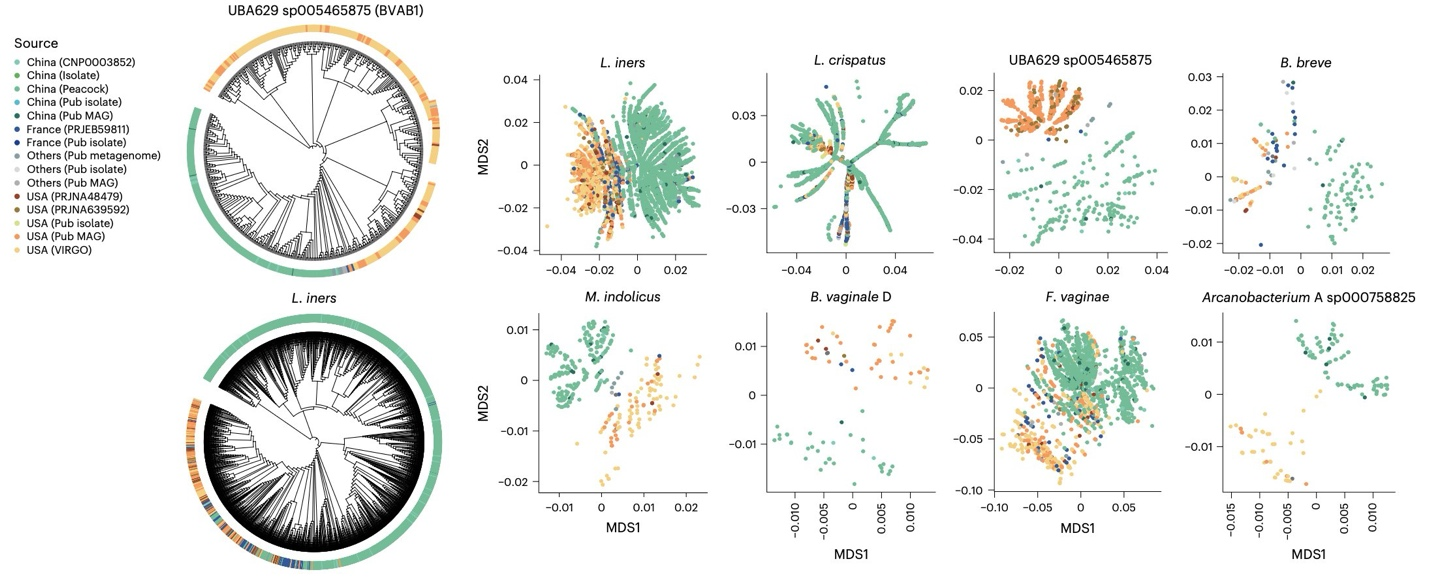 Population-specific genomic variation at the strain level, showing phylogenetic and ordination analyses that reveal distinct clustering of Chinese and American strains across multiple vaginal bacterial species including BVAB1.
Population-specific genomic variation at the strain level, showing phylogenetic and ordination analyses that reveal distinct clustering of Chinese and American strains across multiple vaginal bacterial species including BVAB1.
Analysis of the vaginal virome revealed distinct phage-bacteria co-occurrence patterns across health states. In healthy reproductive-age women, Lactobacillus species and their corresponding phages were co-enriched, whereas under conditions such as HPV infection, BV and menopause, phages were predominantly associated with non-Lactobacillus pathobionts. These ecological patterns suggest phenotype-specific phage-bacteria dynamics, although the underlying mechanisms remain to be elucidated.
Vaginal metagenomic samples contain a high proportion of host DNA (typically over 90%), the researchers leveraged this feature to extract host whole-genome data and microbial profiles from the exact same samples. Applying metagenome-genome-wide association analysis to more than 6,800 women across three independent cohorts, they identified seven host genetic loci that reached study-wide significance and were replicated in at least one validation cohort. The most robust association linked the host opioid receptor gene OPRK1 to the potential pathogen Ureaplasma urealyticum, a microbe implicated in urogenital infections. These findings point to a measurable role for host genetics in shaping vaginal microbial composition, though environmental factors remain the predominant influence and further experimental work is needed to clarify causal mechanisms.
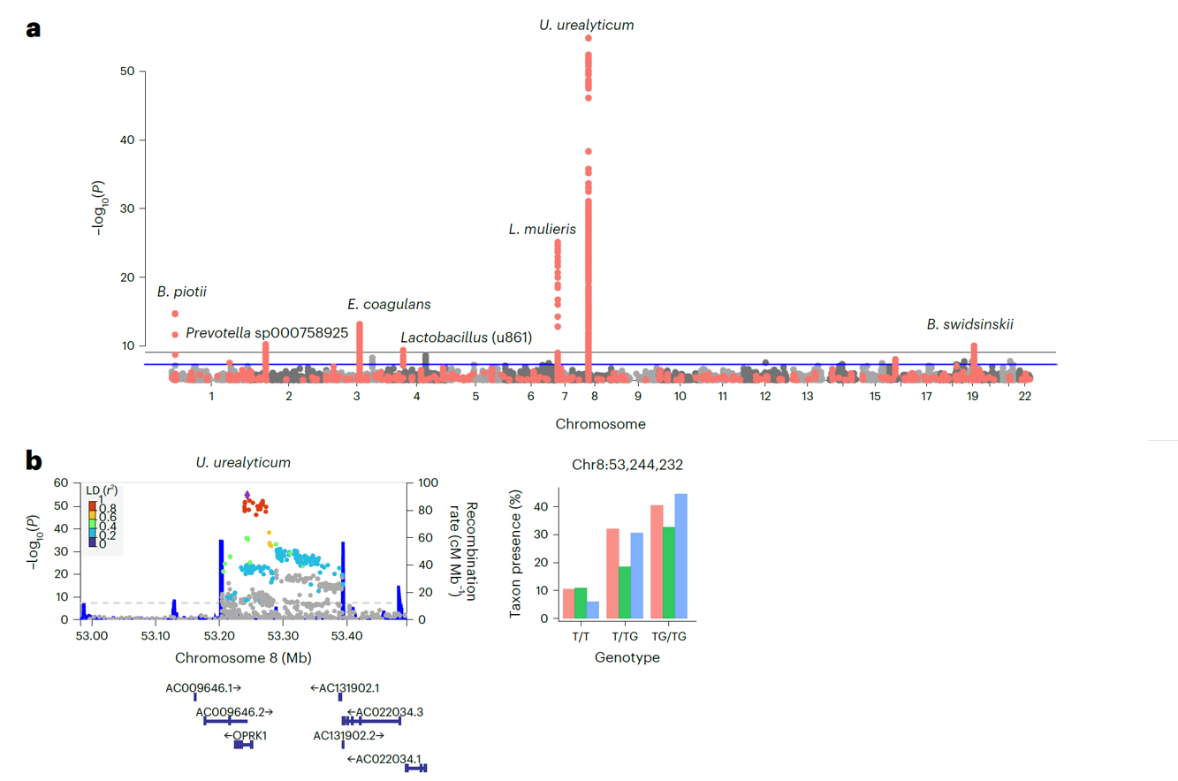 Seven genomic loci reached the study-wide significance from the primary discovery M-GWAS and at least one validation cohort (a), and the strongest and replicated association between OPRK1 and Ureaplasma urealyticum (b).
Seven genomic loci reached the study-wide significance from the primary discovery M-GWAS and at least one validation cohort (a), and the strongest and replicated association between OPRK1 and Ureaplasma urealyticum (b).
Dr. Chen Chen of BGI-Research, one of the study's corresponding authors, noted that GVMG fills a critical gap for Asian populations and provides a foundation for deeper understanding of how vaginal microbes and host genetics jointly shape reproductive health.
By bridging bacteria, viruses, fungi and host genetics across diverse populations, the catalog positions future research to move beyond describing vaginal microbial communities toward understanding how strain-level variation, phage ecology and host genetic background converge on reproductive health outcomes.
All human studies received institutional ethics approval, and all participants provided written informed consent.
This research can be accessed at https://doi.org/10.1038/s41588-026-02639-2