



An open-source foundation model that reads the genomes of human-associated microbes and converts them into reusable numerical signatures is now publicly available. Described in a recent bioRxiv preprint, Genos-m, developed by BGI-Research and Zhejiang Lab, places individual genes, whole genomes, and entire metagenomic samples into a single unified representational framework, producing compact vectors that downstream tools can compare, classify, and search. This cross-scale approach provides an enabling layer for microbiome science that complements today's taxonomy- and annotation-based methods.
 Genos-m converts human-associated microbial genes, genomes, and whole metagenomic samples into reusable numerical signatures, providing a sequence-derived layer that complements taxonomy-based microbiome analysis.
Genos-m converts human-associated microbial genes, genomes, and whole metagenomic samples into reusable numerical signatures, providing a sequence-derived layer that complements taxonomy-based microbiome analysis.
Genos-m extends Genos, the multi-billion-parameter human-genome foundation model that BGI-Research and Zhejiang Lab jointly launched in October 2025. Where Genos targets inherited human genomic variation, Genos-m shifts the same modelling framework to the dynamic, highly diverse sequence space of human-associated microbes. The premise is that general-purpose DNA models are trained for broad cross-species coverage, yet the human microbiome's most informative differences often lie below the species label, in strain-specific genes, gene order, and sequences swapped in from other microbes.
To capture that diversity, the team assembled a training corpus of roughly 1.2 trillion nucleotide tokens spanning 69,056 microbial species. The data paired public reference genomes and bacteriophages with high-quality in-house human-gut metagenome-assembled genomes that contributed about a third of the total.
Architecturally, Genos-m uses a Mixture-of-Experts Transformer with 32 specialist sub-networks, of which only the two most relevant activate for each DNA letter, so that about 330 million of the model's 4.7 billion parameters fire at a time. Working at single-base resolution and supporting context windows of up to one million base pairs, Genos-m can read through multi-gene regions, mobile genetic elements, and most bacteriophage genomes in a single pass. This makes it unusually practical to run: roughly 200-kilobase fragments fit on a 24 GB consumer graphics card, while a single 80 GB GPU handles the full million-base context.
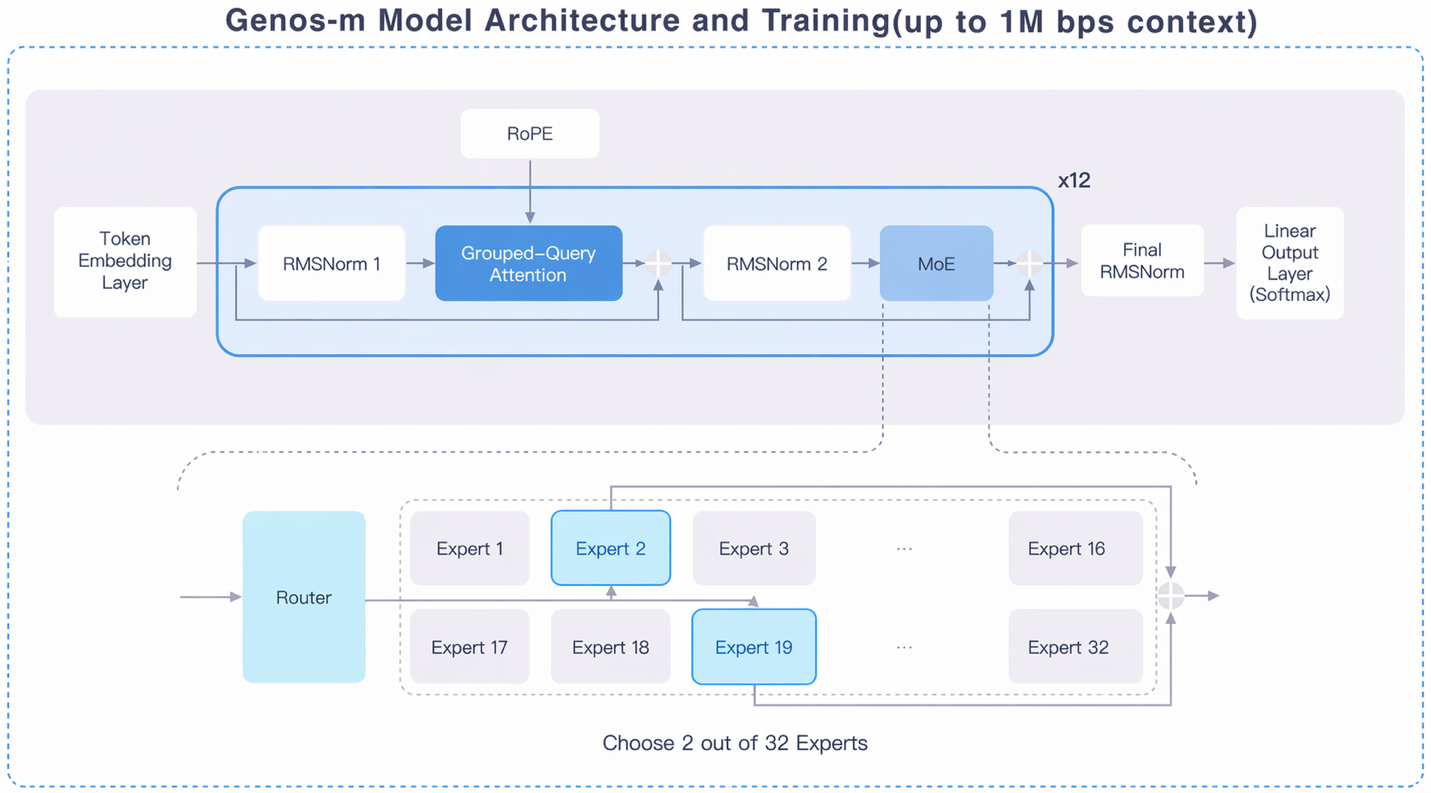 Genos-m's sparse Mixture-of-Experts design (32 experts, Top-2 routing, up to one-million-base context) activates only ~330 million of 4.7 billion parameters per step, keeping long microbial sequences tractable on modest hardware.
Genos-m's sparse Mixture-of-Experts design (32 experts, Top-2 routing, up to one-million-base context) activates only ~330 million of 4.7 billion parameters per step, keeping long microbial sequences tractable on modest hardware.
With the pretrained backbone kept frozen, the team evaluated Genos-m across tasks ranging from short regulatory elements to whole genomes. The model scored highest among the compared models on antibiotic-resistance-gene identification, with an AUROC of 0.9896 on a scale where 1.0 is perfect, and on classifying biosynthetic gene clusters that produce natural antibiotics and other metabolites, at 0.9216. It also led in five of eight gene-fitness prediction tasks that measure how individual genes affect bacterial survival under different conditions. On several other benchmarks it was comparable to, or second behind, the far larger Evo2-40B model, and whole-genome-derived features approached protein-based models on several phenotype labels. These results suggest that domain-focused pretraining can yield competitive representations for human-associated microbial tasks, even with far fewer activated parameters than much larger general-purpose DNA models. Using sparse autoencoders to decompose the model's learned patterns, the team found internal features corresponding to known genomic landmarks such as protein-coding regions and RNA genes, an early step toward interpretability.
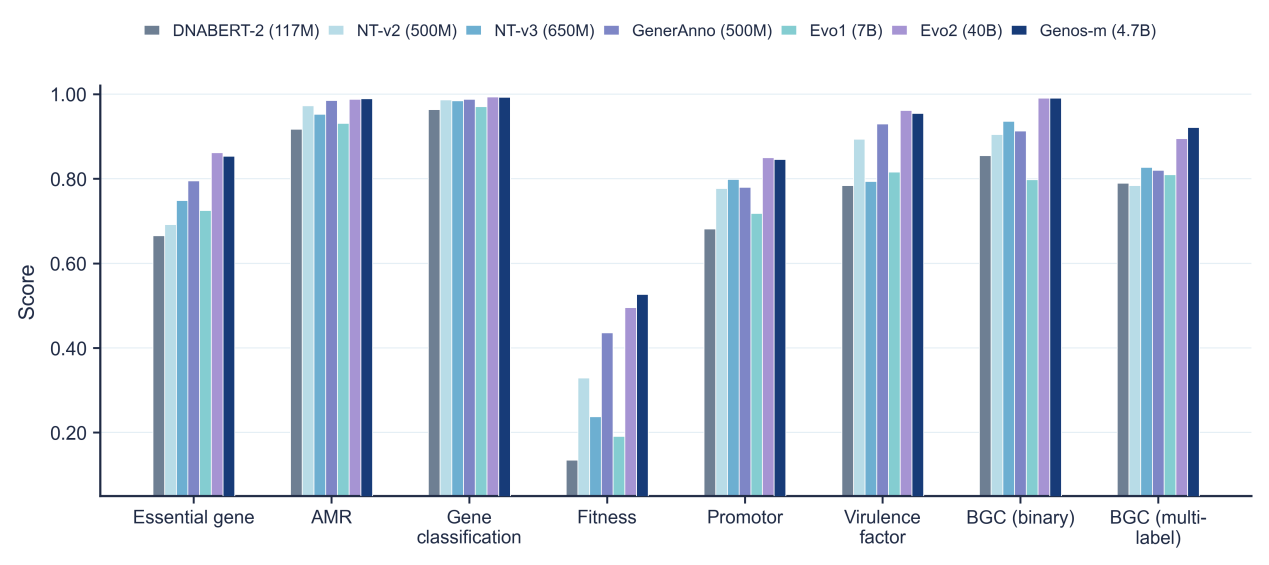 Genos-m ranks among the leading models across antibiotic-resistance, biosynthetic-gene-cluster, gene-fitness, and other benchmarks, posting the best result on several despite far fewer active parameters, showing that targeted pretraining can offset raw scale.
Genos-m ranks among the leading models across antibiotic-resistance, biosynthetic-gene-cluster, gene-fitness, and other benchmarks, posting the best result on several despite far fewer active parameters, showing that targeted pretraining can offset raw scale.
Conventional microbiome analysis relies on tallying which species are present and how abundant they are. The team asked whether incorporating what each species' genome encodes could sharpen disease-related signals, and put Genos-m to work in two gut-metagenome settings. In the first, genome-informed species representations derived from Genos-m-generated genome-level embeddings were fed into a community-level microbiome model trained on about 400,000 unlabeled human gut samples, then evaluated on colorectal cancer case-control classification across 14 independent cohorts. Incorporating the genome-derived information improved mean AUROC from 0.86 to 0.89 within cohorts and from 0.72 to 0.77 when transferring between them — a research-cohort classification benchmark suggesting that disease-associated microbial signals may reside not only in which species are present but in their genomic background.
In the second setting, Genos-m generated stable sample-level embeddings directly from as few as 10,000 sequencing reads. At that ultra-low depth, a linear classifier on the model's output distinguished a sample's geographic origin with an AUC of 0.998, and unsupervised clustering recovered the broad gut-community groupings known as enterotypes with 86% agreement with full-depth profiles, against 54% for a conventional abundance-based method at the same depth. Such low-input characterizations, the authors note, could open a lightweight path for cohort stratification, and quality control where full-depth sequencing is impractical, though they do not replace standard taxonomic profiling.
The team frames Genos-m as adaptable infrastructure and envisions applications in pathogen resistance and virulence identification, probiotic screening and functional evaluation, microbiome assessment and population stratification, and personalized microbiome management. Model weights, inference code, and documentation are openly available under the Apache 2.0 license through BGI-HangzhouAI repositories on GitHub and Hugging Face, and the team invites community testing and contributions.
This research can be accessed at https://doi.org/10.64898/2026.05.21.726868.